| Course ID | Course | Professor | Time | Location |
| Organische Chemie | - |
Organische Chemie
Kurzübersicht

Der Kohlenstoff
Das wichtigste Element der organischen Chemie ist der Kohlenstoff, C. Eine Verbindung die keinen Kohlenstoff enthält, kann keine organische Verbindung sein.Ohne Kohlenstoff wäre die belebte Natur undenkbar. Es gäbe weder Pflanzen, Tiere, Menschen noch Mikroorganismen wie Einzeller, Bakterien und Viren.
In der Natur findet man den Kohlenstoff nicht nur in Gesteinen wie Kalk, CaCO3, und Dolomit, sondern auch elementar als Graphit und Diamant.

CaCO3, Schrattenkalk in den Kurfirsten, Toggenburg, St. Gallen, Schweiz
ÜBUNG: Wenn Kalk, CaCO3 mit Salzsäure, HCl versetzt wird entweicht rauchendes Kohlendioxid, CO2, und zurück bleibt Kalziumchlorid, CaCl2, ein häufig verwendetes Trocknungsmittel in der präparativen Chemie. (Füllstoff des Trockenrohres). Wie lautet die Stöchiometrie:
Antwort:
CaCO3 + 2HCl ===> H2O + CO2 + CaCl2
Graphit und Diamant.
Es sind diese beiden Grund- Modifikationen, die ursprünglich bekannt sind.

Graphit: Dieser besteht aus wabenförmigen Schichten aus Sechsecken. Es sind dehydrierte Benzolringe. Der Kohlenstoff ist im Graphit mit 3 anderen Kohlenstoffatomen verbunden. Das vierte Bindungselektron ist auf der entsprechenden Schichtfäche delokalisiert. Die Schichten werden bloss durch schwache londonsche Kräfte oder Van der Waals-Kräfte zusammengehalten, weshalb sie sich gegenseitig verschieben lassen. Graphit leitet längs der Flächen den elektrischen Strom und wird industriell als Schmiermittel verwendet.

Elementarzelle Diamant. Diese Modifikation ist mit 4 weiteren Kohlenstoffatomen kovalent verbunden. Die Grundzelle besteht aus 3 Sesselförmigen 6-Ringen. Diamant ist das härteste Element (Härte 10) und findet in der Chemie kaum Zugang. Diamant kann, da keine freie, nichtbindende Elektronen vorhanden sind, den elektrischen Strom nicht leiten.
Ein 3. Grund-Modifikation wurde 1985 von Curl, Smalley und Kroto entdeckt.
Das Fulleren

Es handelt sich um einen kugelförmigen Kluster aus Fünf- und Sechsecken.
Das Fulleren muss aus 12 isolierten Fünfecken bestehen, dies verlangt eine geometrisch mathematische Regel nach Euler. Die Anzahl Sechsecke kann beliebig sein.
Der kleinste mögliche Kluster besteht aus 12 fünf- und 20 Sechsecken (C60)
Doktoranden in Tucson, Arizona, sollen Fullerene im Russ nachgewiesen haben.
Die Kohlenstoffatome sind nur 3-bindig. Die übrigen Nicht-Bindungselektronen sind teilweise delokalisiert.
Im Gegensatz zur Diamantmodifikation sind Fullerene löslich und für die Chemie leicht zugänglich.
Weitere Kohlenstoffe, die elementar vorliegen sind: Russ und Koks.
Werden biologische Materialien unter Ausschluss von Luft erhitzt, ensteht Kohle aus Russ und Koks.
Nomenklatur
In einigen Universitäten wird die Nomenklatur eher unterschwellig behandelt.
In Mittel- und Berufsschulen, Gymnasien und Highschools kann die Nomenklatur gar Prüfungsstoff sein. Daher ein Thema, das man nicht vernachlässigen sollte.
Hier werden nur die essentiellsten Regeln vorgestellt. Viele Verbindungen haben jedoch Trivialnamen, die sich soweit eingebürgert haben, dass sie kaum noch wegzudenken sind. Wer im Restaurant nach dem Kochsalz fragt, wird kaum von Natriumchlorid sprechen oder für den Salat wird nicht die Ethansäure sondern den Essig verlangt. Beim hochprozentigen Schnaps spricht niemand das Ethanol an.
Auch der Schweisser arbeitet mit Acetylen (“nicht” mit Ethin) H-C=C-H
Nehmen Sie die Tafel oben zu hilfe. Die Regeln richten sich nach IUPAC (International Union of Pure and Applied Chemistry)
Die essentiellsten Regeln:
- Bestimme die Alkangruppe ( Alk-an, Alk -en, Alk-in, Alkahol, Carbonsäure, Aldehyd, Keton, Amin, Amid etc, siehe Tafel oben).
- Suche die längste durchgehende Kohlenstoffkette und bestimme das organische Stamm-Molekül.
- Substituenten, seien dies organische Reste (siehe unten) wie Alkyl, (Methyl, Ethyl, propyl, butyl phenyl etc) oder anorganische Elemente oder Verbindungen wie Halogenide, Metalle oder einfach andere Elemente oder Verbindungen werden alphabetisch geordnet.
- Stelle der Substituenten haben kleinstmögliche Ziffern.
Beispiele: CH3-CH2-CH2-CH2Cl
Regel 1: Alkangruppe ist ein Alkan
Regel 2: Kette besteht aus 4 gesättigten Kohlenstoffatomen, 4C, = Butan
Regel 3: Subsituent ist Cl, = Chlorbutan
Regel 4: 1-Chlorbutan (aber nicht 4-Chlorbutan)
CH3-CHBr-CH2=CH2Cl
Regel 1: = Alken
Regel 2: = Buten
Regel 3: = Brom, Chlor (alphabetisch ordnen!)
Regel 4: = 3-Brom-1-chlor-1-buten, (aber nicht 4-chlor-2-brom-4-buten)
Sind in einer Verbindung mehrfach dieselben Substituenten oder Mehrfachbindungen vorhanden, werden diese mit griechische Zahlen benannt:
CH2=CH-CH=CH-CH3
1,3-Pentadien
HCCl3 = Trichlormethan
Die Reste: Organische Substituenten enden auf yl.
CH3- Methyl
CH3CH2- Ethyl
CH2=CH2- Ethenyl
(CH3)2CH- i-propyl, isopropyl,
CH3(CH2)3- Butyl
(CH3)2CH-CH2- i-Butyl
C6H5- phenyl
Weitere Substituenten (Reste)
-NH2 amino
-ROH alkoxy
-RCOOH carboxy
CH3O- Methoxy
CH3CH2O- Ethoxy
S- Thio
SCH3- Methylthio
Hier noch ein komplizirteres Beispiel:
Essentielle Aminosäure: Methionin
CH3-S-CH2CH2-CHNH2-COOH
Regel 1: Alkangruppe ist Carbonsäure oder Alkansäure
Regel 2: Stamm-Molekül ist Butansäure
Regel3: Substituenten sind alphabetisch geordnet: NH2 = amino, SCH3 = methylthio
Regel4: Zählen ab COOH = 1, Amino = 2, SCH3 = 4
Diese Verbindung ist chiral und wie alle übrigen Aminosäuren der belebten Natur linksdrehend,(= S, wie sinistra), Das Chiraltätszentrum liegt an zweiter Stelle wie die Aminogruppe, daher 2S. Mehr über Stereochemie im nächsten Kapitel.
Systematischer Name nach IUPAC:
(2S)-Amino-4-(methylthio)butansäure
Methionin
Nomenklatur von cyclischen Verbindungen und Benzolen
Cyclische Verbindungen enthalten einfach die Vorsilbe Cyclo. z.B. 1-Chlor-cyclobutan
Nomenklatur von Benzolen siehe folgende Tafel:

Bei den nächsten Kapitel, bei der cis-, trans-, Isomerie, kommen wir auf die Nomenklatur nochmals kurz zurück.
Isomerie
Haben 2 Verbindungen gleicher Summenformel einen unterschiedlichen Aufbau, spricht man von Isomere.
Wenn die Atome zweier Verbindungen mit gleicher Summenformel unterschiedlich verknüpft sind, handelt es sich um Konstitutionsisomere.
Beispiel.die Summenformel lautet C3H6. Mögliche Konstitutionsisomere sind Cyclopropan und Propen, CH2==CH—CH3.
Organische Moleküle, die in bezug auf Verdrehung eine Veränderung ihrer räumlichen Anordnung erfahren, nennt man Konformationsisomere. Mehr dazu im nächsten Subkapitel.
In der Komplexchemie trifft man bei Koordinationsisomeren unter anderem auch auf Ionisationsisomere.
Haben 2 Verbindungen dieselbe Summenformel und auch dieselbe Verknüpfung aber eine unterschiedliche räumliche Anordnung, spricht man von Stereoisomere.
In der Komplexchemie wird auch, zwischen Hydratisomerie, Koordinationsisomerie und Bindungsisomerie unterschieden, nebst der Stereochemie.
Auf letzteres wird hier in bezug auf die organische Chemie etwas näher eingegangen:
Stereoisomere
Es gibt zwei Arten:
- Diastereomere
- Enantiomere
Bei Diastereomeren liegt der Unterschied bei der Symmetrie. Wie oben erläutert, beide Verbindungen haben wiederum dieselbe Summenformel, beide sind jedoch symmetrisch. Unterscheiden sich jedoch in der räumlichen Anordnung.
Wichtig, beide Verbindungen haben unterschiedliche chemische und physikalische Eigenschaften. Solche Verbindungen werden mit Präfix cis- und trans- oder Z und E(wenn ungesättigt, vgl nächstes Kapitel)) benannt.
Bei Enantiomeren haben beide Substanzen mit gleicher Summenformel keine Symmetrie, unterscheiden sich wie die linke und rechte Hand oder unprofessionel ausgedrückt sind spiegelbild-isometrisch. Im Gegensatz zu Diastereomeren haben Enantionmere dieselben physikalischen Eigenschaften, mit einer Ausnahme. Bestrahlt man das eine Enantionmer mit linear polarisiertem Licht erfährt die Ebene des Lichtstrahls eine Drehung nach rechts oder links. Daraus entstand die von Cahn, Ingold und Prelog vorgeschlagene Nomenklatur. (R)-, = Rechtsdrehend, (S) Linksdrehend, (R= rectus, S = sinister). In der Biochemie bei den Zuckern findet man auch die älteren Kürzel D/L, dextro/laevo = rechts/links, haben aber nicht ganz dieselbe Bedeutung.
Die assymmetrische Verbindung wird als optisch aktiv oder chiral bezeichnet. Eine Verbindung die ein Symmetriezentrum oder eine Symmetrieebene (auch Spiegelebene genannt)
hat, ist achiral, wenn beide fehlen ist sie chiral. Zudem lassen sich symmetrische Substanzen zur Deckung bringen.
Hier sind einige Beispiele des Alltags: Liegenschaften:

Dieses Landhaus hat auch auf der linken Seite eine Laube und dieselben Eingänge auch die linke Dachseite ist mit einem Dachfenster versehen. Die hintere Seite ist ebenfalls symmetrisch auch wenn das Muster ein anderes ist. Dieses Bauernhaus hat somit in der Mitte dem Dachfirst entlang eine Symmetrieebene (Spiegelebene) und ist somit achiral.

Diese zwei Wohnhäuser haben weder eine Symmetrieebene noch ein Symmetriezentrum. Sie sind deshalb chiral. Beide Wohnhäuser stehen “spiegelbildlich” zueinander. Men kann sie als Enantiomere bezeichnen.
Es folgen einige Verbindungen als Kalottenmodelle: Wasserstoff = weiss, Kohlenstoff = schwarz, Chlor = grün und Sauerstoff = rot

Diastereomere: cis-1,2-dichlorethen und trans-1,2-dichlorethen

Sind das Enantiomere?

Nein! sie sind identisch 1,3-Dichlorpropan

Sind aber das Enantiomere?

richtig! Sie lassen sich nicht in Deckung bringen. R- und S-trans-1,2-Dichlorcyclopropan

2 Symmetrieebenen: Dichlormethan achiral

1,4-cyclohexandiol, hat ein Symmetriezentrum, deshalb achiral

Wie findet man heraus ob eine chirale Verbindung rechts oder linksdrehend ist?
Zuerst muss das Chiralitätszentrum ermittelt werden. In der organischen Chemie ist das meistens ein C-Atom. Eine organische Verbindung kann mehrere Chiralitätszentren haben, so bei den Zuckern. Es werden hier nur Verbindungen mit 1 Chiralitätszentrum behandelt.
Danach werden die 4 an das C gebundenen Substituenten nach bestimmten Prioritätsregeln, gemäss Cahn, Ingold und Prelog, geordnet. Es werden hier nur die 2 Wichtigsten erwähnt. Wir machen das mithilfe der Milchsäure. Systematischer Name: 2-Hydroxypropansäure.

- Priorität hat das Atom mit der höheren Ordnungszahl. O = 16 kommt vor N = 14. Bei Isotopen hat das Atom mit der höheren Masse Priorität.
- Betrachtet wird das Atom der ersten Sphäre, das direkt an das C-Atom bzw an das Atom des Chiralitätszentrum gebunden ist. Sind mehrere gleichartige Atome in der 1. Sphäre vorhanden, werden die Atome der 2. Sphäre entsprechend geordnet. Enthält ein Substituent Doppel- oder Dreifachbindungen, wird es wie wenn 2 oder 3 Atome vorhanden wären, behandelt. C==O wird zu O—C—O, C==C wird zu C—C—C.
Milchsäure: Priorität: 1. OH, 2. COOH, 3. CH3, 4. H

Wir haben nun die Prioritäten bestimmt und können nun feststellen ob die Verbindung rechts- oder linksdrehend ist. Falls ein Kalottenmodell vorhanden ist, muss dieses so aufgestellt werden, dass das Atom mit der letzten Priorität, hier das H-Atom vom Betrachter am weitesten entfernt ist. Sind die ersten drei Substituenten 1 > 2 > 3 im Uhrzeigersinn angeordnet, dann ist Verbindung rechtsdrehend und wird mit Präfix (R)- bezeichnet. Präfix (S) erhält das linksdrehende Enantiomer.
Wenn keine Kalottenmodelle vorhanden sind oder erstellt werden können, finden die Fischer-Projektionen Anwendung. Man zeichnet ein Kreuz, wobei die Vertikale die Hauptkohlenstoffkette kennzeichnet. Zuoberst ist derjenige Substituent, bei welchem nach der systematischen Nomenklatur die Numerierung beginnt. Hier wäre das die Gruppe COOH. Entsprechend wie oben beschrieben, kann man auch hier herausfinden, ob die Verbindung links- oder rechtsdrehend ist.
Mesomerie
In Mittelschulen, Highschool und Abiturklassen, taucht auch der Begriff Mesomerie auf. Manche Moleküle eben in der organischen Chemie können nicht korrekt dargestellt werden. Es wird angenommen, dass die negative Ladung oder auch die Elektronenpaare bei Mehrfachbindungen auf mehrere Atome verteilt sind. Beispiel: Benzol und 1,3-Pentadien.
Aber auch in der anorganischen Chemie.
Im Kapitel, Moleküle siehe oben: Hier sind die Strukturformeln der Kohlensäure, H2CO3 und der Salpetersäure, HNO3, wiedergegeben.

2 mal deprotoniert, formuliert man das Carbonat-Anion, CO3(-II) in 3 verschiedenen Grenzformeln. Das Molekül ist planar und die Landung, -2, kann nicht präzis geortet werden. Die Ladung ist delokalisiert.
Auch für die letztere Verbindung HNO3 gibt es 2 mögliche Grenzformeln, weil man nicht weiss, liegt die negative Ladung, -I, beim oberen oder unteren Sauerstoff.
In der organischen Chemie wimmelt es von mesomeren Strukturen. In Universitäten und in höheren Lehrbüchern der organischen
Chemie kommt das Wort Mesomerie kaum vor. Man spricht die Verbindung sei resonanzstabilisiert.
Achtung!
Summenformel C5H8
CH2==CH–CH2–CH==CH2 und CH2==CH–CH==CH–CH3 sind weder Mesomere, noch Enantiomere noch Diastereomere, sondern lediglich Konstitutionsisomere!
Die Kohlenwasserstoffe
Kohlenwasserstoffe verstehen sich als Verbindungen von Kohlenstoff, C, und Wasserstoff, H.
Grundsätzlich wird zwischen
- Gesättigten
- Ungesättigten
Kohlenwasserstoffe unterschieden.
Gesättigte Kohlenwasserstoffe enthalten nur Einfachbindungen. So sind Methan, Propan oder auch Dimethylpropan (Neopentan) aber auch Cyclohexan (Siehe Bild zuoberst) oder Bicyclo(4,4,0)decan (Decalin) und Bicyclo(2,2,1)heptan (Norbornan) gesättigte Kohlenwasserstoffe. Man nennt sie Alkane.

Ungesättigte Kohlenwasserstoffe haben Doppel oder Dreifachbindungen.
Der Hauptunterschied zwischen Einfach- und Doppel- oder Dreifachbindung liegt jedoch an der Bewegungsmöglichkeit. Die C—C-Bindung lässt sich gegenseitig verdrehen.
Die räumliche Anordnung in bezug auf die unterschiedlichen Stellungen durch Verdrehung und Verzweigung nennt man Konformation. Moleküle mit unterschiedlicher Konformation sind Konformationsisomere.
Beim Ethan drehen die Methylgruppen, -CH3, bei Raumtemperatur um die C—C-Bindung, von der gestaffelten zur ekliptischen Konformation und zurück zur Gestaffelten. Letztere ist energetisch begünstigt, denn bei der ekliptischen Konformation stossen sich die beiden gegenüberstehenden H-Atome gegenseitig ab.

Ethan gestaffelte Konformation

Ethan ekliptische Konformation
Beim Cyclohexan, (siehe beide Bilder unten) wechselt das Molekül dauernd von der Sessel- über die Twist- zur Wannenform und zurück. Die Sesselform ist aber energetisch begünstigt.

Cyclohexan, Wannen-Konformation

Cyclohexan, Twist-Konformation

Cyclohexan, Wannen-Konformation
Beim Ethen, CH2==CH2, gestattet die Doppelbindung keine Drehung. (gilt auch für Dreifachbindungen!) Das Molekül ist deshalb planar, bzw liegt auf einer Ebene. Ebenso ist Benzol, das 3 Doppelbindungen hat planar und kennt im Gegensatz zum Cyclohexan weder eine Sessel- noch eine Wannenform. Dies hat jedoch auch andere Gründe, dazu mehr weiter unten.
Ungesättigte Kohlenwasserstoffe mit ein oder mehreren Doppelbindungen nennt man Alkene:
Beispiele
Propen, 2-Methyl-1,3-butadien, (Isopren = ein Terpen),a-Pinen (Hauptbestandteil von Terpentin), b-Carotin (Provitamin A).Limonen Cyclooctatetraen, Benzol, Azulen.
(letztere 2 Verbindungen, Benzol und Azulen, sind Aromate)
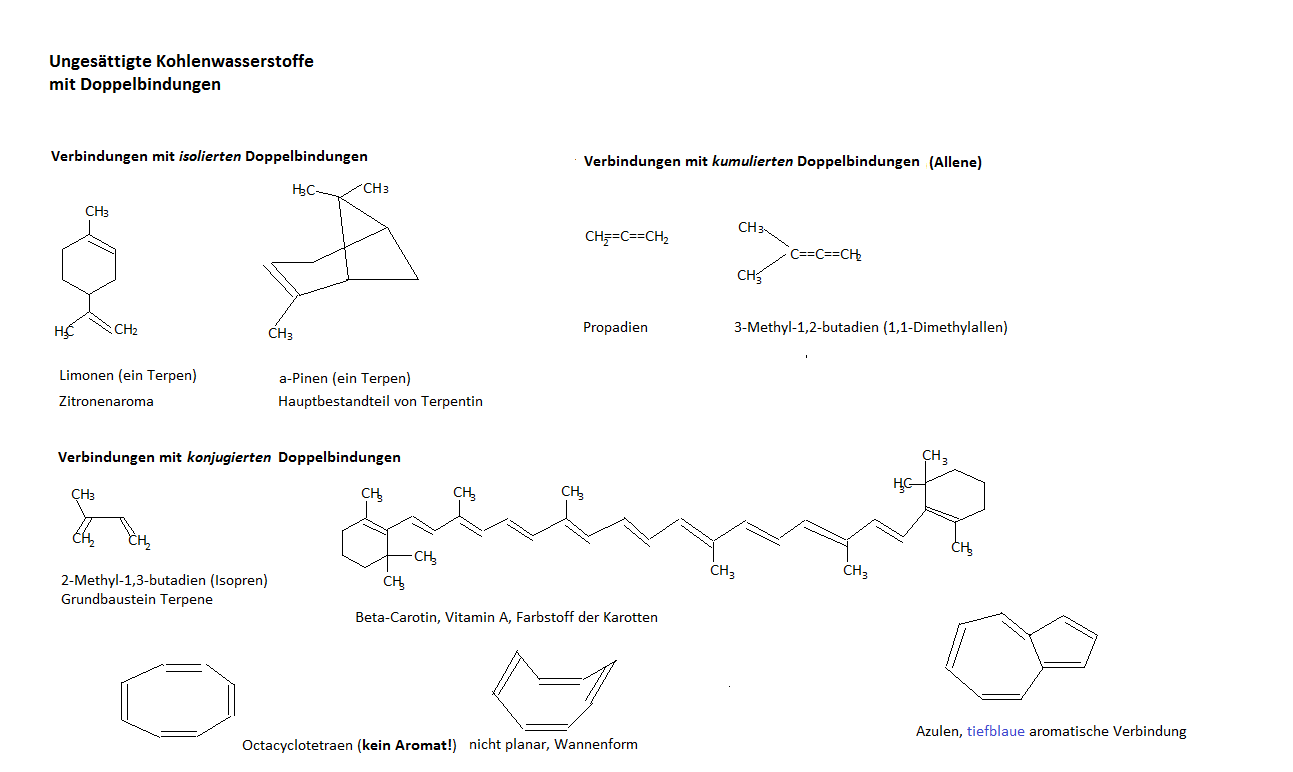
Kohlenwasserstoffe mit mehreren Doppelbindungen werden auch Polyene genannt. Jenachdem ob zwischen 2 Doppelbindungen eine, mehrere oder keine Einfachbindungen vorhanden sind, wird zwischen konjugierten, isolierten und kumulierten Doppelbindungen unterschieden. Butadien und Benzol sind konjugierte Kohlenwasserstoffe und sind resonanzstabilisiert (oder mesomer vgl Isomerie)
Ungesättigte mit ein oder mehreren Dreifachbindungen werden Alkine genannt.
Ethin, (Acetylen), 2-Butin, Butadiin, Benz-in (sehr instabil)

Benz-in, Dehydrobenzol, ist ein Aromat, aber sehr instabil (vgl. nächstes Subkapitel)
Die H-Atome, direkt an die C mit Dreifachbindungen gebunden, sind schwach sauer und lassen sich mit starken Basen wie NaNH2 deprotonieren.
Aromatische Kohlenwasserstoffe und Hückel-Regel
Das Wort Aromat geht auf einige in Pflanzen enthaltene aromatisch riechende Substanzen zurück. Tatsächlich handelt es sich dabei um Benzol-Verbindungen wie z.B. Coffein und Vanillin.
Aromatische Kohlenwasserstoffe sind Ringe oder Mehrfachringe mit konjugierten Doppelbindungen. Der wichtigste Vertreter ist das Benzol.
Aromatisch ist eine Verbindung wenn,
- die Struktur einen Ring darstellt.
- es Konjugierte Doppelbindungen im gesamten Ring hat. Einfachbindungen dürfen nicht nacheinander erfolgen.
- sie Planar ist (eine Ebene darstellt)
Fehlt eines dieser Eigenschaften ist es kein Aromat.
Damit die aromatische Verbindung auf einer Ebene liegt, müssen die Elektronen der Doppelbindungen gleichmässig verteilt (delokalisiert) sein. Dessen Ladungsdichte muss sich senkrecht über oder unter der Ringebene befinden, damit sich die Ecken nicht bewegen können.
Aromatisch sind nebst Benzol auch Mehrfachringe oder mehrkernige Benzole wie Naphthalin, Anthracen und Phenanthren.
Auch Ringe mit mehr oder weniger als 6 Kohlenstoffatome mit konjugierten Doppelbindungen sind aromatisch, fass obgenannte Regeln erfüllt sind.
Beispiele: Azulen, (Bild oben), 1,2,5,7,9,11,13,15,17-Cyclooctadecanonaen. (vgl. Zeichnungen oben konjugierte Doppelbindungen).
Nun sind die 3 obgenannten Regeln nur beschränkt anwendbar.
Cyclobutadien und 1,3,5,7-Cyclooctatetraen sind kein Aromaten, weshalb?
Letztere Verbindung haben wir bereits oben bei den konjugierten Doppelbindungen kurz behandelt: 1,3,5,7-Cyclooctatetraen ist nicht planar, es beschreibt eine Wanne.
Beim Cyclobutadien sind alle der 3 obgenannten Regeln erfüllt, trotzdem ist es kein Aromat. Ausserdem ist Cyclobutadien äusserst instabil und zersetzt sich bereits bei -200°C.
Wann ist die Verbindung aromatisch?
Hückel Regel
Eine einfache mathematische Fromel verrät, ob die Verbindung aromatisch ist oder nicht.
Denn eine cyclische Verbindung mit konjugierten Doppelbindungen besitzt 4n + 2 Doppelbindungselektronen. Benzol hat 3 konjugierte Doppelbindungen, 6 zusätzliche Elektronen sind dazu notwendig.
Die Gleichung lautet
4n + 2 = 6
Wir lösen nach n auf und erhalten
n = 4/4 = 1, eine natürliche ganze Zahl ohne Dezimalstelle oder Brüche. Benzol ist also ein Aromat. Dieselbe Lösung liefert auch das Benz-in. Das Elektronenpaar der 3. Bindung der Dreifachbindung wird nicht mitgezählt, ist aber dafür verantwortlich, dass der Ring verzerrt ist. Die Molekülorbitaltheorie wird in höheren Lektionen behandelt.
Wie sieht dies nun bei Cyclobutadien aus. 4 zusätzliche Elektronen für die konjugierten Doppelbindungen sind notwendig.
Die Gleichung lautet:
4n + 2 = 4
Lösung nach n ergibt
n = 2/4 = ½ odere 0,5. nun haben wir einen Bruch oder eine Dezimalstelle, deshalb ist Cyclobutadien kein Aromat.

Dasselbe gilt auch für 1,3,5,7-Cyclooctatetraen mit 8 Doppelbindungselektronen.
4n + 2 = 8, n = 6/4 = 1,5, kein Aromat wie oben bereits erwähnt.
Azulen besitzt auf beiden Ringen insgesamt 10 Doppelbindungselektronen.
4n + 2 = 10, n = 8/4 = 2 = Aromat
Nebst aromatischen Kohlenwasserstoffe können auch Heterocyclen aromatisch sein. Pyridin ist ein einfaches Beispiel. Achtung: Das Elektronenpaar auf dem Stickstoff, liegt in der Ringebene und trägt nicht dazu bei.
Im Gegensatz dazu liegt das Elektronenpaar beim Pyrrol senkrecht zur Ringebene und gilt als Elektronenpaar, das zum aromatischen Charakter beiträgt. Die Hückel-Gleichung ist dieselbe wie beim Benzol.
Nichtaromatische Ringe, die Regel 2 erfüllen kann man mittels Lewis-Basen oder – Säuren aromatisch machen, das Molekül ist jedoch nicht mehr ladungsneutral.
Reaktionen und Reaktionsmechanismen der organischen Chemie
In diesem Subkapitel wird Grundsätzliches über die Reaktionen in der organischen Chemie vorgestellt.
Vorweg ist festzuhalten, dass die meisten Reaktionen Gleichgewichtsreaktkionen sind, und dass oft mesomere (resonanzstabilisierte) Übergangszustände durchlaufen werden.
Grundsätzliches zum Reaktionsmechanismus:
Es beschreibt:
- Sämtliche Reaktionsgleichungen und Übergangszustände die zum Produkt führen.
- Der Pfeil zeigt immer an, wo das Elektronenpaar hingeht.
Die organische Chemie gliedert sich in vier Reaktionstypen:
(Gemäss Peter Sykes, Reaktionsmechanismen der Organischen Chemie)
- Substitutionen
- Additionen
- Eliminierungen
- Umlagerungen und Verschiebungen
Hier werden
- Die Markownikoff Addition
- Die nucleophile Substitution und die Eliminierung
- Die elektrophile Substitution am Benzolring
- Die Oxidation
- Der Addition-/Eliminierungs-Mechanismus
- Die Polymerisation und die radikale Substitution
kurz behandelt.
Es wird empfohlen die Übersicht organischer Verbindungen und die Tabelle der Elektronegativitäten bereitzuhalten. Wichtig zu wissen ist, dass Sauerstoff, O2, elektronegativer als Stickstoff, N2 und N2 elektronegativer ist als Kohlenstoff, O < N < C.
Vorerst wird etwas Grundsätzliches zu den Carbenium-Ionen gesagt: Wird einem Kohlenwasserstoff ein Atom samt Bindungselektronenpaar (z.B. ein Chlorid-Anion, Cl-) entfernt entsteht ein Carbokation. Man unterscheidet zwischen
- CH3+, methyl-Kation
- RCH2+, primäres Carbokation
- R2CH+, sekundäres Carbokation
- R3C+, tertiäres Carbokation
Wichtig: Die Stabilität nimmt bei den Carbokationen in dieser Reihenfolge von 1 bis 4 zu.
Grund dafür ist, dass sich C—C-Bindungen leichter polarisieren lassen als C—H-Bindungen.
Und ergänzungshalber sei erwähnt, dass umgekehrt die Stabilität von Carbanionen von 1 – 4 abnimmt. H3C- > RH2C- > RHC- > R3C-.
R ist ein beliebige Atom (von H verschieden) oder Alkylrest.
Markownikov Addition
Wenn Propen mit Wasserstoffbromid umgesetzt wird, erhält man 90 % 2-Brompropan. Der Mechanismus läuft dabei wie folgt ab: Das Proton, das hier die Funktion als Lewis-Säure bzw als Elektronenpaar-Akzeptor ausübt, setzt sich bevorzugt am Ende der Doppelbindung
fest. folglich entsteht das stabilere sekundäre Carbokation, insbensondere weil das Proton weniger elektronegativ ist als das Bromid-Ion.
CH3—CH==CH2 + HBr → (CH3—CH+—CH3) + Br-
Das sekundäre Carbenium-Ion (sekundäres Carbokation) wird bevorzugt gebildet, weil es stabiler ist.
Das elektronegativere Bromid-Ion nimmt daher Position 2 ein und man erhält somit überwiegend 2-Brompropan.
Anti-Markownikov Addition. Es ist auch möglich, vorwiegend 1-Brompropan herzustellen. Dies Reaktion läuft jedoch nach einem Radikalmechanismus und wird in höheren Kursen behandelt.
Nucleophile Substitution
Bei der nucleophilen Substitution in der organischen Chemie wird am Kohlenstoffatom ein gebundenes Atom oder eine Gruppe durch eine andere ersetzt. Nucleophil bedeutet soviel wie kernliebend (nucleus = Kern). Das noch an das C gebundene Atom oder die Gruppe nennt man Abgangsgruppe. Das neue an den Kohlenstoff zu bindende Atom oder die neue Gruppe nennt man Nucleophil.
Je nach Stabilität des Carbokations, gibt es 2 Grenzfälle von Reaktionen:
Direkte Beispiele:
NaOH + CH3Cl ===> H—O —— CH3 —— Cl ===> H—O—CH3
Wenn aus Chlormethan Methanol hergestellt wird, entsteht vorerst als Übergangszustand ein aktivierter Komplex. Dabei wird das C Atom von hinten bzw der vom Chlor abgewannten Seite vom Hydroxid-Ion, OH-, das hier als Lewis-Base wirkt, angegriffen. Beim sich im Gleichgewicht befindenden aktivierten Komplex werden die drei H-Atome umgeklappt (invertiert). Man stelle sich einen Regenschirm bei starkem Wind vor. Die Bindung des abtretenden Chlorid- Ions wird geschwächt wird nach erfolgter Umklappung der 3 H-Atomen gelöst. Dies ist die nucleophile Substitution 2. Ordnung, denn der geschwindigkeitsbestimmende Schritt ist der aktivierte Komplex. Die Reaktion verläuft, da es sich um ein instabiles Carbokation handelt, bimolekular. Die Abkürzung dazu lautet Sn2.

weiss = H, schwarz = C, grün = Cl, rot = O
Anders verläuft die Reaktion, wenn am C-Atom anstellen von 3 H, 3 Methylgruppen gebunden sind. Beim 2-Chlor-2-methylpropan oder t-Butylchlorid trifft das Hydroxid-Ion auf eine Verzweigung von 3 Methylgruppen und wird dabei solange gehindert, bis das Chlorid-Ion abgeht. Dabei entsteht ein relativ stabiles tertiäres Carbokation, das dann rasch vom Hydroxid-Ion von hinten oder von vorne angegriffen wird.

Der geschwindigkeitsbestimmende Schritt ist hier die abtretende Gruppe, hier das Chlorid-Ion. Die Reaktion läuft unimolekular, man spricht von einer Sn1-Reaktion.

Eliminierung
Wir bleiben gleich beim t-Butylchlorid dessen Chlor-Atom sich soeben entfernt hat. Nun verzichtet das Hydroxid-Ion auf den Angriff auf das tertiäre Carbokation und entfernt dafür ein Proton einer Methylgruppe.Reultat: 2-Methylpropen. Es handelt sich um den E1-Mechanismus, die ebenso unimolekular verläuft. Sehr gute Ausbeuten sind zu erreichen, wenn dieselbe Reaktion in wässrigem Ethanol* oder mit einer anderen schwachen Base ausgeführt wird.
Bei der bimolekuaren Eliminierung, E2, können starke Basen im Überschuss beim 2-Chlor-2-methylpropan einer Methylgruppe sofort ein Proton entfernen, noch bevor das Chlorid-Ion abgegangen ist.

Weil aber die Bindung des Chlorid-Ions dadurch geschwächt wird, läuft die Reaktion bimolekular bzw konzertiert oder gleichzeitig ab. Der geschwindigkeitsbestimmende Schritt ist wie bei der Sn2-Reaktion der aktivierte Komplex.
Zur Abgangsgruppe. Gute Abgangsgruppen sind Halogenide wie Chlor, Brom und Iod,
nicht jedoch Fluor, F.
*Für Chemische Synthesen im Labor sollte nicht Brennsprit verwendet werden. Dieser Alkohol ist mit etwas Pyridin versetzt, damit er nicht getrunken werden kann.
Die elektrophile Substitution am Benzolring (Erstsubstitution)
Bei der nucleophilen Substitution haben wir erfahren, dass die angreifende Verbindung ein Nucloephil sein muss und dass es sich dabei um eine Lewis-Base also um einen Elektronenpaar-Donator handeln muss. Der Angriffspunkt ist der Nucleus oder ein Atomkern. Meistens ist es in der organischen Chemie ein Kohlenstoffatom, weil es in der Regel am wenigsten elektronegativ ist. Es wird bei Additions/Eliminierungs-Mechanismus weiter unten nochmals darauf eingegangen.
Ganz das Gegenteil trifft zu, wenn am Aromat, ein H-Atom durch einen Substituenten ersetzt wird. Hier handelt es sich um eine elektrophile Substitution. Das angreifende Atom oder Verbindung ist ein Elektrophil und wirkt als Lewis-Säure, (Elektronenpaar-Akzeptor). Das Elektronenpaar entnimmt das Elektrophil dem Benzolring.
Intermezzo
Vorerst wird hier noch kurz das Thema Katalysatoren behandelt. Ein Katalysator ist ein Atom oder eine Verbindung, die die Reaktion beschleunigt, ohne dabei verbraucht zu werden. Die Reaktion kommt dabei mit weniger Reaktionwärme aus.
Es gibt 2 Arten von Katalysatoren:
- Homogene Katalysator
- Heterogene Katalysator
Der wesentliche Unterschied besteht darin, dass der homogene Katalysator am Reaktionsprozess teilnimmt und dabei vorübergehend auch Bindungen eingeht. Diese finden bei der elektrophilen Substitution an Aromaten anwendung. AlCl3, FeBr3, H2SO4 sind wichtige homogene Katalysatoren.
Beim heterogenen Katalysator sind es oft elementare Übergangsmetalle mit Spuren von anderen Elementen, die Reaktanden absorbieren. Das heisst, Die Reaktanden berühren bloss die Oberfläche des katalysierenden Metalls, gehen jedoch keine Bindungen damit ein. Eisen, Fe, Platin, Pt, Nickel und Platin/Rhodium, Pt/Rh sind heterogene Katalysatoren. Letzterer, Pt/Rh, ist Bestandteil des Abgaskatalysators, der Stickstoffoxid, NO und Kohlenmonoxid, CO zu untoxischen Gasen wie Kohlendioxid, CO2 und Stickstoff, N2 rückführt:
NO + CO → N2 + CO2
Beispiel, die Hydrierung (H-Addition) von Alkenen mit Übergangsmetallen als Katalysator, z.B. Platin, Pt, Palladium, Pd oder auch Nickel, Ni. Wenn die Oberfläche mit Wasserstoff, H2, berührt wird, löst sich die Bindung von H2. Beide H-Atome werden kurz an die Metalloberfläche gebunden ehe sie an das Alken addiert werden. Auch das Alken kommt mit einem seiner Doppelbindungen mit der Metalloberfläche kurz in Berührung.
Wenn Sie organisch chemische Reaktionsversuche mit Metallen im Labor ausführen, sollten Sie nicht Metallpulver sondern Metallspäne verwenden, wegen der grösseren Oberfläche.
In der biologischen Chemie nehmen Enzyme und Coenzyme (Vitamine) die Rolle von Katalysatoren ein.
Benzoladdition mit homogenen Katalysator, was passiert dabei genau?
Wir stellen Toluol aus Benzol her. Dazu verwenden wir Chlormethan, CH3Cl und Aluminiumtrichlorid, AlCl3 als Katalysator.(Friedel-Crafts-Alkylierung). Die Gesamtreaktion lautet:

AlCl3 wirkt als Lewis-Säure (Elektronenpaar-Akzeptor) und entnimmt CH3Cl ein Chloratom.
CH3Cl + AlCl3 → CH3+ + AlCl4-
Das ist die Katalysator-Reaktion
CH3+ ist sehr kurzlebig und bildet nun als starke elektrophile Gruppe vorerst mit Benzol einen Komplex. EDP/EPA-Komplex. Das primäre Carbokation, CH3+ lagert sich an ein bestimmtes Ringkohlenstoffatom an. Dabei entsteht ein mesomeres Benzenium- Ion.

Hier bleiben CH3 und H noch gebunden, ehe das H-Atom vom einem Chlorid-Anion von AlCl4+ abgetrennt wird. Die Schlussreaktion:

Dabei wird der Katalysator AlCl3 rückgebildet und es entsteht Salzsäure, HCl.
Die Katalysator-Reaktionen lauten für die anderen Katalysatoren, FeBr3 und H2SO4 ähnlich. Das Elektronphil, das ein Kation sein muss, ist kurzlebig und sehr reaktiv.
Brom-Addition, Herstellung von Brombenzol
Br2 + FeBr3 = Br+ + FeBr4-
Brom wird heterolytisch gespalten zu Br+ und Br-, ersteres agiert als Elektrophil und letzteres lagert sich an FeBr3 an, analog wie bei AlCl3
Herstellung von Nitrobenzol mit H2SO4 als Katalysator
HNO3 + 2 H2SO4 =è NO2+ + H3O+ + 2HSO4-
NO2+ ist das Elektrophil
Herstellung von Benzolsulfonsäure, ein essentielle Verbindung in der präparativen Chemie.
SO3 + H2SO4 =è SO3H+ + HSO4-
Schwefeltrioxid wird protoniert und elektrophil gemacht: SO3H+
Zweitsubstitution am Benzolring
Soeben wurde am Benzolring ein H-Atom durch einen elektrophilen Substituenten ersetzt.
Ein weiterer Substituent soll nun eingeführt werden. Das Verfahren läuft ähnlich ab wie bei der Erstsubstitution. Die Frage ist nur: An welchem Ring-C-Atom setzt sich der Zweitsubstituent fest? Das hängt allein davon ab, inwiefern sich die Elektronendichte über dem Benzolring durch den Erstsubstituenten verändert. Die Elektronendichte wird erhöht durch Substituenten mit freien Elektronenpaaren. Massgebend ist das erste an den Ringkohlenstoff gebundene Atom. Dabei spielt auch die Elektronegativität eine wichtige Rolle.
Allgemein gilt: Substituenten, die ein oder mehrere freie Elektronenpaare besitzen, erhöhen die Elektronendichte am Benzolring.
Beispiel: Zweitsubstitution an Methoxy-Benzol, Nitrierung mit HNO3

Die Halogenide, F, Cl, Br, I, tragen etwas weniger zur Elektronendichte bei, da sie sehr elektronegativ sind.
Der 2. Substituent setzt sich an Position 2, ortho-Stellung oder Position 4, para-Stellung fest.
Substituenten die kein freies Elektronenpaar besitzen und mit einem elektronegativeren Atom gebunden sind, verringern die Elektronendichte, indem diese abgezogen wird.
Beim Nitrobenzol mit NO2 als Substituent besitztder Stickstoff nun kein freies Elektronenpaar und ist mit 2 O-Atomen gebunden, die elektronegativer sind als Stickstoff. N = 3, O = 3,4. Das N-Atom hat gar eine positive Formalladung.
Die Reaktion läuft langsamer ab.
Die Elektronendichte wird verringert bzw Richtung Substituent abgezogen, weil das direkt an den Benzolring gebundene Atom kein freies Elektronenpaar besitzt und die anderen gebundenen Atome elektronegativer sind. Folgende Erstsubstituenten zwingen den zweiten Substituent zur Metastellung.
Beispiel: Zweitsubstitution der Benzoesäure mit Brom.

Anstelle von Überschussladungen entstehen an denselben Stellen nun Elektronenlücken. Diese positiv geladenen Stellen, zwingen das 2. Elektrophil, das auch positiv geladen ist, eine andere Position einzunehmen. Der 2. Substituent setzt sich somit an Position 3 oder 5 fest, Meta-Stellung. Indem man sämtliche mesomeren Grenzstrukturen aufzeichnet, lässt sich die Metastellung herleiten. Dazu mehr in höheren Kursen.
Die Zweitsubstitution von Toluol und generel von Alkylbenzol stellt ein Spezialfall dar. Der Zweitsubstituent nimmt Ortho- oder Para-Stellung ein, obwohl das C-Atom kein freies Elektronenpaar besitzt. Mit unserem bisher erworbenen Kenntnissen kann man dies zeigen, indem sämtliche mesomeren Strukturen simulativ aufgezeichnet werden:
Nur bei der ortho- und para-Stellung entsteht u.a. eine mesomere Übergangsstruktur: Ein tertiäres Carbenium-Ion, ein Carbo-Kation.

Nur bei der ortho- und para-Stellung entsteht u.a. eine mesomere Übergangsstruktur: Ein tertiäres Carbenium-Ion, ein Carbo-Kation.
Aus der oben beschriebenen Markownikov-Addition wissen wir, dass tertiäre Carbokationen stabiler sind als sekundäre. Die Nitrogruppe nimmt Ortho- oder Para-Stellung ein.
Die Nitrogruppe ist stärker elektronenziehend als die Methylgruppe. Deshalb werden 2 weitere NO2-Gruppen in Meta-Stellung problemlos am Aromatenring substituiert. Man erhält schliesslich Trinitrotoluol. Dieses Pulver dient als Sprengstoff in Handgranaten. Es lässt sich relativ leicht im Labor herstellen.
Oxidation und Reduktion in der organischen Chemie
Es wird empfohlen das Kapitel 7, Oxidation und Reduktion nochmals zu nachzuschlagen.
Unter Oxidation versteht man ursprünglich Reaktion mit Sauerstoff. Das ist eher eine alchimistische Definition. Wir nehmen gleich ein Beispiel: Wir verbrennen Methan.
CH4 + O2 → CO2 + H2O. Das ist eine exotherme Reaktion. Es werden dabei 800 kj/mol an Energie frei.

Wir ermitteln nun die Oxidationszahlen beim Kohlenstoffatom. Dabei arbeite man mit dem Schema der Elektronegativitäten und stellen fest:
H ist mit 2,2 elektropositiver als C (= 2,6) und O (=3,4) ist elektronegativer als C.
Die Oxidationszahlen in der organischen Chemie werden bestimmt, indem die Bindungselektronen dem elektronegativeren Atom zugeteilt werden. Die Oxidationszahl ergibt sich aus der Differenz zwischen
- Total Bindungselektronen plus allfälligen nichtbindenden Elektronenpaare minus Valenzelektronen.
Methan: C hat 8 Bindungselektronen. Davon werden die 4 Valenzelektronen von C abgezogen. 8 – 4 = 4, es verbleiben 4 elektronen (also -IV). Wasserstoffatome haben je die Oxidationszahl +1 (+I). Da Methan ladungsneutral ist muss die Summe sämtlicher Oxidationszahlen der gesamten Verbindung null ergeben.
-IV + 4*(+I) oder -4 + 4 = 0.
Am Ende der Reaktion existiert die Kohlenstoffverbindung aus CO2. Hier besitzen die Sauerstoffatome eine höhere Eletronegativität (=3,4), weshalb ihnen die Bindungselektronen zugeteilt werden. Die Oxidationszahlen betragen je O-Atom Anzahl Bindungselektronen plus 2 nichtbindende Elektronenpaare (=4) -6 Valenzelektronen = -2. Für das C-Atom beträgt die Oxidationszahl +4. Kontrolle:
2*-2 + 4 = 0
CO2 ist wie das Methan ladungsneutral. Der Kohlenstoff hat somit insgesamt 8 Elektronen an die beiden Sauerstoffatome abgegeben, ist somit um 8 Elektronen oxidiert.
Aber was passiert genau bei dieser Verbrennung?
Methan wird zuerst zu Methanol, dann entsteht Methanal (Formaldehyd) und fast zuletzt Methansäure (Ameisensäure). Zuletzt wird aus der kleinsten Carbonsäure CO2 abgespalten (Decarboxylierung). Die beiden verbleibenden H-Atome werden kurz zu Wasserstoff, H2, und es enststeht eine Knallgasreaktion mit O2 als letzte Stufe.
H2 + 1/2O2 → H2O

Sauerstoff ist ja im Überfluss vorhanden, sonst wäre es eine Katastrophe. Auf dem Mars könnte man mit gerade mal einem Druck von 6 millibar CO2 weder atmen noch autofahren (ausgenommen Elektro- und Solarfahrzeug) geschweige den mitgebrachtes Holz verbrennen.
Oxidation in der präparativen Chemie geht vom Alkohol aus. Aus einem Alkohol wird ein Aldehyd, ein Keton oder eine Carbonsäure hergestellt. Im Beispiel oben haben wir ja Methanol bereits über die Sn2 Reaktion erhalten. Das Methanol wird nun oxidiert.
Achtung! Methanol ist sehr giftig! Einnahme kann Erblindung verursachen!
Ein sehr essentielles Oxidationsmittel ist Dichromat: K2Cr2O7, Na2Cr2O7 (in H2SO4 = Jones-Reagens) und CrO3 sind im Handel erhältlich. Wenn Methanol mit einem dieser Oxidationsmittel reagiert erhält man direkt Ameisensäure. Will man erreichen, dass die Oxidation beim Formaldehyd gestoppt wird, muss CrO3 mit Pyridin komplexiert werden.
Mit CrO3(pyridin)2 können praktisch alle Akohole zu Aldehyden oder Ketonen synthetisiert werden. Es ist schwieriger Carbonsäuren aus Ketonen zu erhalten, weil dabei eine C-C-Bindung aufgebrochen werden muss. Ein wichtiges Oxidationsmittel in der organischen Chemie ist auch Kaliumpermanganat, KMnO4.
Oxidationsmittel in der Medizin:
Wie in Kapitel 7 bereits erwähnt, kann man den Blutzucker im Urin mit Silbernitrat, AgNO2, nachweisen. (Tollens-Nachweis) Glucose ist eine Aldose und lässt sich gut in Carbonsäure überführen, dabei wird Silber reduziert und elementares Silber scheidet aus und setzt sich an der Gefässwand fest. Ein Silberspiegel bildet sich aus.
Ein weiteres in der Medizin oft verwendetes Reagens ist die Fehlingprobe. Diese besteht aus Kupfer(II)-hydroxid, Cu(OH)2 und bildet zusammen mit basischer Tartrat-Lösung (Alkalisches Salz der Weinsäure) eine hellblaue Komplexverbindung. Bei zuckerpositiven Werten wird Kupfer(II) zu Kupfer(I) reduziert. (Nimmt ein Elektron auf). CuO2 entsteht und die Farbe wechselt von hellblau zu gelbrot.
Reduktion in der organischen Chemie
Im Gegensatz zu Oxidationen lassen sich Carbonsäuren, Aldehyde und Ketone zu Alkoholen reduzieren.
Wichtig zu wissen: Die Carbonylgruppe ist polarisiert. Das lässt sich auch mit Oxidationszahlen erklären. Die insgesamt 8 bindenden und nichtbindenden Elektronen werden dem Sauerstoffatom zugeordnet. Das sieht man auch an der Resonanzstruktur.

Beim C- Atome greifen nur Nukleophile an. Beim O-Atom können sich somit nur Elektrophile festsetzen. Meistens ist das ein Proton. Mehr dazu erfahren Sie weiter unten
Zwei essentielle Reduktionsmittel:
- Natriumborhydrid, (Natriumtetrahydridoboranat), NaBH4
- Lithiumaluminiumhydrid, (Lithiumtetrahydroalanat), LiAlH4
Bei diesen beiden häufig verwendeten Reduktionsmittel wird ein Hydrid-Ion freigesetzt.
Achtung: das Hydrid-Ion ist kein Proton! Es ist ein Wasserstoffatom mit einem Elektronenpaar.
Wenn beide Verbindungen in Wasser gelöst werden, entsteht Wasserstoff, H2, und Hydroxid, OH-. Das Hydrid-Ion, H–, des Reduktionsmittel verbindet sich mit einem Proton des Wassers.
NaBH4 + 4H-O-H → NaOH + B(OH)3 + 4 H-H
LiAlH4 + 4H-O-H → LiAl(OH)4 + 4 H-H
LiAlH4 ist reaktiver als NaBH4. Die Synthese mit LiAlH4 sollte möglichst wasserfrei in Diethylether, CH3CH2OCH2CH3, durchgeführt werden. NaBH4 kann in wässrigem Ethanol gelöst werden.
Bei Arbeit mit diesen beiden Reduktionsmittel ist besondere Vorsicht geboten.
Die Synthese darf nur in einem staatlich zugelassenen Labor durchgeführt werden. Die zu reduzierende Verbindung in Ether gelöst muss mit Trockeneis, Eis oder Flüssigstickstoff vorerst gekühlt werden, damit die Reaktion nicht zu heftig ausfällt. Tropfenweise ist das Reduktionsmittel in der Kapelle mit einer Spritze dem Reaktionsgemisch beizufügen. Das Kapellenschiebefenster muss halbgesenkt sein. Hände sind mit Gummihandschuhe zu schützen. Schutzbrille ist ebenfalls Pflicht.
Reaktionsmechanismus vereinfacht.
Aus Acetaldehyd wird Ethanol reduziert.
Mit NaBH4 in Ethanol
NaBH4 + CH3CHO → CH3CH2OH + (Na+)H3BOCH2CH3

Es können nun 3 weitere Ethanal-Moleküle angegriffen werden. Wenn alle Hydrid-Anionen verbraucht sind, verbleiben neben 4 Ethanol-Molekülen, Natrium-Tetraethoxyborat, (Na+)B(OCH2CH3)4-.
Mit LiAlH4 greift ebenfalls ein Hydrid-Ion das C-Atom der Carbonyl-Gruppe an. Das Sauerstoff-Atom der Carbonylgruppe setzt sich an der Stelle des Aluminium fest, wo sich soeben ein Hydrid-Ion freigesetzt hat. Dieser Prozess wiederholt sich bis alle 4 Hydrid-Ionen am Aluminium durch die Carbonylgruppen der Acetaldehyd-Moleküle ersetzt sind.

Durch Reaktion mit Wasser, (Hydrolyse) entstehen 4 Moleküle Ethanol sowie Lithium- und Aluminiumhydroxid. (vgl Bild oben)
LiAlH4 ist sehr reaktiv. Carbonsäuren können direkt zu Alkoholen reduziert werden.
Aus Essigsäure, CH3COOH, kann damit Ethanol, CH3CH2OH, hergestellt werden.
Allerdings kann die Reaktion, wie die Erfahrung zeigt, zum Stillstand kommen. Dabei fällt oft Lithiumethoxylat, CH3CO2-Li+, aus.
Um dies zu verhindern, stellt man über einen Additions/Eliminierungs-Mechanismus einen Ester her. Dazu mehr im nächtsten Subkapitel.
Wolff Kishner Reduktion (Eine Namensreaktion)
Ketone werden zu Alkane reduziert. Die Carbonylgruppe wird eliminiert.
Reduktion von Cyclohexanon zu cyclohexan:

Man braucht dazu Hydrazin, N2H4, (85% in Wasser gelöst) und Natronlauge, NaOH.
Das Cyclohexanon ist in einem Alkoholether, OHCH2CH2OCH2CH2OH (Sdp 245°) gelöst. Die Hydrazin-Lösung wird beigegeben und das Ganze mit NaOH erhitzt. Danach wird die Mischung mit Wasser aufbereitet und man erhält Cyclohexan.
Der Addition-/Eliminierungs-Mechanismus
Das Proton, das bei einem Aldehy z.B, beim Acetaldehyd am C-Atom der Carbonylgruppe gebunden ist, ist kaum abzutrennen oder zu ersetzen. Man kann auch sagen, Wasserstoff ist eine miserable Abgangsgruppe. Nur die Oxidation macht aus dem Aldehyd eine Carbonsäure. Aus Acetaldehyd entsteht Essigsäure.
CH3CHO → CH3COOH

Als Reagens dienen die Oxidationsmittel K2Cr2O7, Na2Cr2O7, CrO3, was soeben im letzten Subkapitel behandelt wurde.
Anstelle des H-Atoms haben wir nun eine Hydroxid-Gruppe, eine relativ gute Abgangsgruppe,welche sich leichter substituieren lässt.
Die geschieht über den Additions-/Eliminierungsmechanismus. Allgemein kann dieser wie folgt dargestellt werden:

L ist die Abgangsgruppe, X das Nukleophil.
Mit dem Additions-/Eliminierungsmechanismus lassen sich zahlreiche Synthesen in der organischen Chemie herleiten:
- Die Synthese von Säurechlorid: CH3COOH → CH3COCl mit Thionylchlorid, SOCl2 oder Phosphorpentachlorid, PCl5.
- Die Synthese eines Amids, RCONH2 aus Säurechlorid, CH3COCl
- Die Estersynthese z.B. Ethanol und Essigsäure reagieren mit einer Säure als Katalysator zu Essigsäureethylester (Ethylacetat) oder Nagellack. Mechanismus siehe Zeichnung unten.
- Die Esterhydrolyse. (Esterverseifung) aus Essigsäureethylester entsteht als Rückreaktion wiederum Ethanol und Essigsäure. Katalysator kann auch eine Base sein.
- Die Claisen-Kondensation. Aus Essigsäureethylester entsteht Acetessigester.
- Die Aldolkondensation oder Aldoladdition. Dies ist lediglich eine Additions-Reaktion von 2 Aldehyden. Keine Eliminierung findet statt. Der Mechanismus ist jedoch ähnlich. Siehe Zeichnung unten.
Synthese von Essigsäureethylester (Ethylacetat) säurekatalysiert
Vereinfachter Mechanismus:
Der Angriff von Ethanol verläuft ohne Säure eher reaktionsträge. Das Proton ist elektrophil und setzt sich bevorzugt am Carbonylsauerstoff fest. Dank diesem Proton kann die OH-Gruppe, die Abgangsgruppe, leichter entfernt werden, welche ein zusätzliches Proton des Ethanols erhält: Man spricht von Wasser als Abgangsgruppe. Schlussendlich geht auch das Katalyseproton der ursprünglichen Carbonylgruppe ab. Ethylacetat ist Bestandteil von Nagellack.

Esterhydrolyse, (Verseifung).
Die säurekatalysierte Esterhydrolyse bzw die Rückreaktion der Estersynthese verläuft nach demselben Schema. Am Schluss erhalten Sie Essigsäure und Ethanol zurück.
Es handelt sich hier um typische Gleichgewichtsreaktionen. Um die Gleichgewichte weiter nach rechts zu verschieben ist es ratsam, bei der Veresterung Ethanol und bei der Esterhydrolyse Wasser im Überschuss einzusetzen. Es sollte möglichst reiner Ethanol, (kein Brennsprit) verwendet werden.
Esterhydrolyse, (Verseifung). Aus Essigsäureethylester entsteht basenkatalysiert, z.B. mit NaOH, Carboxylat (die deprotonierte Essigsäure) und Ethanol. Hier greift das Hydroxid-Anion als Nukleophil den Carbonyl-Kohlenstoff an.

Verseifung wird die Esterhydrolyse deshalb genannt, weil bei der Hydrolyse von Fetten (= Glycerinester höherer Carbonsäuren) Seifen hergestellt werden. Nartriumsalze der Fettsäuren sind harte Seifen (Kernseifen), Kaliumsalze sind Schmierseifen.
Aldolkondensation
Synthese 3-Hydroxybutanal
Ausgangsverbindung ist wieder Acetaldehyd. Davon reagieren 2 Moleküle. Es ensteht ein Aldol, ein Aldehyd das auch ein Alkohol ist. Zur Wiederholung: das H-Atom beim C der Carbonylgruppe lässt sich nur durch Oxidation ersetzen. Die Aldolkondensation ist deshalb nur eine Additionsreaktion. Diese wird hier erwähnt, weil auch dieser Mechanismus ähnlich verläuft und um einen kleinen Vorgeschmack zum nächsten Subkapitel, Polymersation, zu geben.
Zuerst ist festzuhalten, dass die Methylgruppe, die ein Carbonylkohlenstoff als Nachbar hat relativ sauer ist. Dort lässt sich mit einer Base, mit NaOH z.B., ein Proton entfernen. Dabei entsteht ein mesomerer Übergangszustand und das Acetaldehyd wird zu einem Nukleophil. Dieses kann wie beim Additions / Eliminierungsmechanismus das C-Atom der Carbonylgruppe des zweiten Acetaldehydmoleküls des angreifen. Der Mechanismus ist derselbe, nur dass wegen diesem fest gebundenen H-Atom am zweiten Acetaldehyd, das zum O-Atom nach oben geschobene Elektronenpaar nicht mehr hinunterkommt, weshalb eine Eliminierung ausbleibt. An dieser Stelle wird das entfernte Methylproton des ersten Acetaldehyds wieder zurückgegeben. Das Hydroxid-Anion, OH-, der homogene Katalysator als Base, wird wieder rückgebildet.

Die Polymerisation und die radikale Substitution
Bleiben wir beim Acetaldehyd. 2 Moleküle hat man soeben reagieren lassen. Daraus entstand ein Aldol (siehe Darstellung oben). Das Acetaldehyd bezeichnet man als Monomer. Reagieren beide Moleküle miteinander, entsteht ein Dimer. Nun kann man das Aldol mit einem dritten, vierten und weiteren Molekülen, Acetaldehyd, reagieren lassen, dann haben wir ein Polymer.
A1 + A2 + A3 +… Ax = A-A-A-A……
Zu den Polymeren zählen Kunststoffe, wie Plastik und Gummi und Naturstoffe, wie Stärke, Cellulose und Proteine. Letztere können aus 20 verschiedenen Aminosäuren bestehen.
Herstellung von Makromolekülen oder Polymeren erfolgen durch folgende Mechanismen.
- Polyaddition
- Polykondensation
- Kationische und anionische Polymerisation
- Radikalische Polymerisation
Die soeben erwähnte Aldolkondensation ist eigentlich eine Polyaddition. Ein komplizierteres Beispiel ist Polyurethan, aus 1,6-Hexandiisocyanat und Ethylenglykol.
Bei der Polykondensation wird pro Verknüpfung ein Äquivalent Wasser, H2O, abgespalten, sie erfolgt über den oben behandelten Additions-/Eliminierungsmechanismus. Produkte sind u.a.
- Nylon 66, ein Polyamid, aus Hexandisäure und 1,6-Diaminohexan.
- Polyethylenterephtalat, PET, ein Polyester, aus Terepththalsäure und Ethylenglykon
PET ist Bestandteil der kaum mehr wegzudenkenden PET-Getränkeflaschen.

Achtung: Benzin kann in PET-Flaschen nur kurz aufbewahrt werden höchstens ca 1 – 2 Monate. Die O-Atome bilden nach 5 – 6 Monaten mit den Wasserstoffatomen des klopffesten Benzins (verzweigte Heptane und Oktane) Wasserstoffbrücken und es gelangt auch etwas des Polymers in Lösung. Das Benzin verbindet sich quasi mit PET. Das Benzin verliert seine physikalischen Eigenschaften wie Flüchtigkeit, und lässt sich nicht mehr zünden. Es entsteht eine eher geruchlose, farblose, Flüssigkeit mit einer erhöhten Oberflächenspannung. (Es bilden sich schöne runde Tropfen)
Bei der ionischen Polymerisation sind Lewis-Säuren (kationische Polymerisation) und Lewis-Basen (anionische Polymerisation) zuständig. Besonders bedeutungsvoll ist die Polymerisation von Alkenen. Mit einer Lewis-Säure z.B. BF3 entstehen aus Propen Polypropylen. Bedingung ist eine Doppelbindung. Alkane kann man nicht polymerisieren. Die Lewis-Säure ist ein Elektronenpaar-Akzeptor wie wir wissen. Somit kann BF3 seine Elektronenlücke mit einem Elektronenpaar der Doppelbindung eines Alkens füllen. Das Alken mutiert zu einem Carbenium-Kation. Wir wissen auch, dass das die positive Ladung am 2. C-Atom des Propens vorhanden sein muss, weil es ein sekundäres Carbo-Kation und somit stabiler ist. An dieser Stelle lässt sich nun ein weiteres Propen-Molekül anbringen. Das Carbon-Kation pflanzt sich fort bis zum Kettenabbruch.
Mit der anionischen Polymerisation kann man Carbonylverbindungen zu polymerisieren. Es entstehen Polyether. Als Startersubstanz dient ein Anion wie z.B. Methoxid-Anion, CH3O-.
Ethen lässt sich einfacher mittels radikalischer Polymerisation synthetisieren, weil primäre Carbenium-Kationen instabiler sind.
Was ist ein Radikal?
Es ist ein Atom oder eine Verbindung mit einem ungepaarten nicht bindenden Elektron und wird mit einem Punkt gekennzeichnet. A•
Chlor, Cl2 (oder Brom, Br2) wird mit Licht oder UV zu 2Cl. gespalten. Wir haben nun 2 Chlor- Radikale. Das Chlor-Radikal kann an Alkanen ein H-Atom ersetzen. Radikale Substitution:
Die Gesamtreaktion
CH3–CH3 + Cl2 === CH3–CHCl + HCl
Sieht der Mechanismus wie folgt aus:
Kettenstart:
Aus Chlorgas entstehen 2 Radikale
Cl2 → 2Cl•
Kettenfortpflanzung:
Es entstehen weitere Radikale. Aus Ethan entsteht ein Ethan-Radikal. Dieses wiederum reagiert mit Chlor. Es entsteht Chlorethan und ein Chlorradikal
Cl• + CH3CH3 → CH3CH2• + HCl
CH3CH2• + Cl2 → CH3CH2Cl + Cl•
Kettenabbruch:
Je 2 Radikale verbinden sich wieder zurück
CH3CH2• + Cl• → CH3CH2Cl
Cl• + Cl• → Cl—Cl
In der organischen Chemie können Alkane direkt in Halogenalkane überführt werden. Im Forschungs-Labor geschieht das mit Halogen-Butanimid, (-Succinimid). Aus N-Brom-Butanimid, (N-Bromsuccinimid, NBS) und Wasserstoffbromid, HBr, entsteht vorerst Br2, das dann mit Licht oder UV in Bromradikale, Br• gespalten wird.
Herstellung Polyethylen:
Die radikale Polymerisation läuft ähnlich ab:
Aus Ethen als Monomer entsteht mithilfe eines Radikals (Radikalstarter) ein Ethenyl-Radikal, das weitere Ethen-Moleküle addieren kann. Dies geschieht bis zum Kettenabbruch, bzw bis keine Ethenmoleküle mehr vorhanden sind.
Übersichtshalber wird angenommen, dass das Polymer nur aus 2 Einheiten besteht, ein Dimer.
Als Radikale dienen hiezu u.a. Peroxide, ROO., oder Azoverbindungen. Radikale sollten nur sparsam eingesetzt werden.
Kettenstart:
Ra2 === 2Ra•
Ra• + CH2==CH2 → RaCH2—CH•
Kettenfortpflanzung:
Ra–CH2—CH• + CH2==CH2 → Ra–CH2—CH2—CH2—CH.
Kettenabbruch:
Ra—CH2—CH2—CH2–CH• + Ra• → Ra—CH2—CH2—CH2—CH2—Ra
Ra• + Ra• → Ra–Ra*
*Am Ende der Polymerisation sollte kein Radikal mehr vorhanden sein. Da hier bloss ein Dimer entsteht, wird angenommen dass noch Radikale vorhanden sind.
Radikale kann man nicht als Katalysatoren bezeichnen. Insbesondere bei der Polymerisation sind Radikale schlussendlich im Endprodukt enthalten.
Kettenstart und Kettenwachstum läuft relativ gut ab, weil Monomere auch zu Peroxiden modifizierten Sauerstoff aus der Luft aufnehmen können.
Wie formuliert man Polymere?
Ethen CH2==CH2 [-CH2—CH2-]x Polyethylen
Weiter Polymere:
Chlorethen CH2==CHCl [-CH2—CHCl-]x Polyvinylchlorid, (PVC)
Fluorethen CF2==CF2 [-CF2—CF2-]x Polytetrafluorethylen, (Teflon, Pfannenbeschichtung)
Ungesättigte Polymere, Herstellung von Kautschuk und Gummi.
Aus Dienen und Alkinen erhält man Polymere, die noch Doppelbindungen enthalten.
In der Natur existiert Kautschuk aus Polyisopren. Synthetisch entsteht Kautschuk durch Polymerisation von Butadien, CH2==CH—CH==CH2. Die noch verbliebene Doppelbindung dieses Polymers,
[-CH2–CH==CH—CH2-] lässt sich weiter vernetzen. Durch Reaktion mit Schwefel, S, Fachausdruck “Vulkanisation” dieses Kautschuks entsteht durch Vernetzung der Ketten untereinander Gummi.

Herstellung Polybutylen, Kautschuk. (Bild oben)

Vulkanisation, aus Kautschuk und Schwefel entsteht Gummi. (Bild oben)
Elementarer Schwefel baut sich sich aus S8 Molekülen auf. Die Struktur ist ein 8-Ring mit 2 Dellen.

In chemischen Reaktionen wird einfachheitshalber nur das Symbol S geschrieben.
Schwefelbrücken spielen auch in der Biochemie eine wesentliche Rolle, bei der Bildung von Proteinen und Polypeptiden.
Schwefel findet man in der Natur als Sulfide von Übergangsmetallen: FeS, Pyrit, PbS, Bleiglanz, ZnS, Zinkblende, und als Erdalkalisulfate: CaSO4, Anhydrit, CaSO4*2H2O, Gips, MgSO4, Magnesiumsulfat und im Erdöl. Mit Schwefel gewinnt man Benzol aus Cyclohexan, weshalb im Erdöl und im Benzin recht viel Benzol vorhanden sind.
Cyclohexan + S → Benzol + H2S
In der Nähe von aktiven Vulkanen entweicht Schwefelwasserstoff, H2S. (Geruch nach faulen Eiern). Dort kann sich auch elementarer Schwefel freisetzen. Grosse Lager elementaren Schwefels befinden sich in den USA (Texas und Louisiana)
Verbrennt man Schwefel ensteht SO2, Schwefeldioxid, das in Wasser gelöst die schweflige Säure, H2SO3, bildet. Aus SO2 entsteht auch Schwefeltrioxid, SO3 und hat in der organischen präparativen Chemie grosse Bedeutung.
Literatur
- K. Peter C. Vollhart, Organische Chemie, VCH Verlag GmbH, D-6940 Weinheim (BRD) 1988, 1990
- Peter Sykes, Reaktionsmechanismen der Organischen Chemie, VCH Verlag GmbH, D-6940 Weinheim (BRD) 1988
- Charles E. Mortimer, Das Basiswissen der Chemie, Georg Thieme Verlag Stuttgart . New York 1987 5. Auflage
© Alle Rechte sind ibusciencecollege.com vorbehalten